western blot 的原理是什么?
刚好最近做完了实验,实验室要带师弟师妹做western blot实验了,western blot实验相关的经验总结写了几个晚上,这里一起分享给大家,刚入门的同学可以多看看~
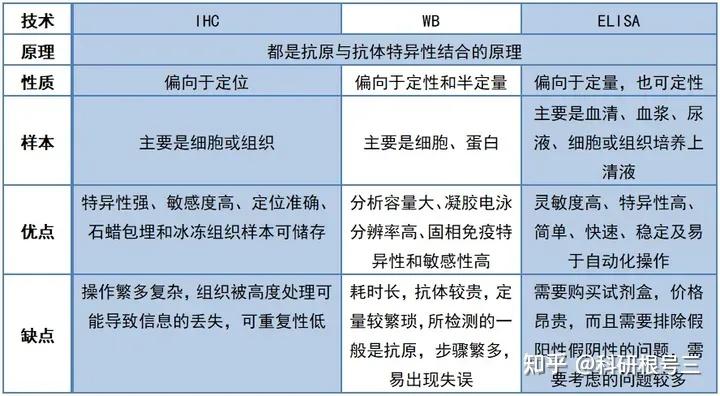
WB,是Western blotting的缩写,即蛋白质印迹法,又称免疫印迹(immunoblotting),也就通过电泳技术分离不同组分,再采用印迹技术将蛋白质转移(“印”)到特定的膜上,再利用抗原-抗体的特异性结合原理,用抗体作为探针去实现检测复杂样品中的靶标蛋白的方法,这项技术也可以用来检测不同时期蛋白表达的水平以及蛋白相互作用等方面。
实验原理和用途
原理:通过电泳区分不同的蛋白组分,并转移至固相支持物,通过特异性抗体作为探针,对靶蛋白进行检测。

用途:定性检测,判断目标蛋白有无表达,目标蛋白分子量大小。常配合ELISA、FACS、IHC、IF等手段使用。
最全的生物实验protocol与生物实验操作视频学习资料
112个科研软件免费版(数据处理+绘图+翻译+生物实验相关等)
下载链接:https://pan.quark.cn/s/34a75dd2241d
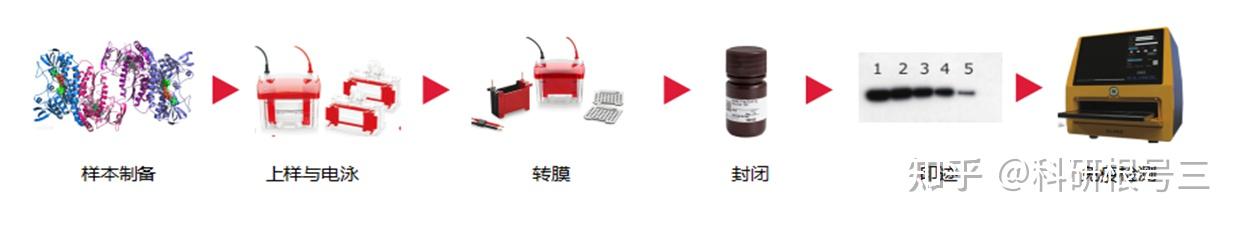
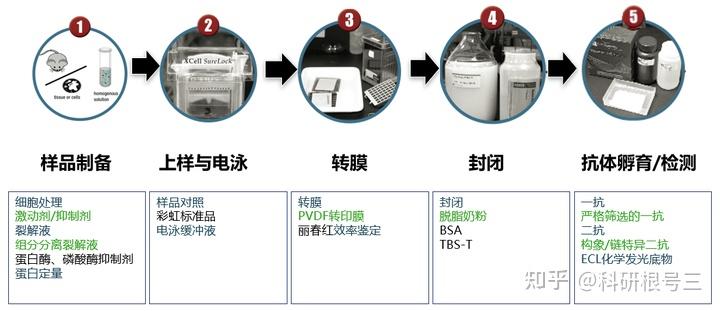
一、Western Blot标准操作流程
样本制备→凝胶电泳→蛋白转膜→封闭→抗体孵育→检测显影
1、样本制备
1)实验前需要清楚的重要信息
- 所用的细胞/组织中表达需要检测的靶标蛋白吗?
- 表达丰度如何?是否在WB的检测范围内?
- 靶蛋白的亚细胞定位是哪里(在核还是在膜或者其它位置)?靶蛋白分子量是多少(分子量决定了电泳胶浓度以及转膜大小)?
- 是否需要特殊刺激才会表达?(有些蛋白无刺激不表达)
- 是否在疾病组织或细胞中才会有表达?
- 是否有多种异构体?是否有多种翻译后修饰形式?(决定抗体的选择)
利用数据库查询重要信息:
Uniprot:http://biogps.org/#goto=welcome
解靶标蛋白基因信息、别称、功能、可变剪切、定位及可修饰类型等
Proteinatlas:https://www.proteinatlas.org/
通过IHC提供基因在正常组织和肿瘤组织中的表达变化和定位
BioGPS:http://biogps.org/#goto=welcome
不同样本中基因的mRNA表达水平和靶标蛋白的表达水平
PAXdb:http://pax-db.org/
不同物种组织和细胞中蛋白丰度
DepMap:https://depmap.org/portal/
肿瘤细胞mRNA表达量
或者也可以在参考文献中找到相关蛋白的信息。
2)样本制备流程
- ① 样本获得:组织或细胞取材
- ② 样品裂解:从组织或者细胞中释放获取蛋白
- ③ 蛋白定量:获悉蛋白浓度,为后续电泳步骤提供上样量依据
- ④ 蛋白变性:使蛋白变性形成线性结构,方便跑胶和抗体结合
处理细胞--抑制剂与激动剂
靶标蛋白可能需要进行刺激才会表达,这时候就需要找到合适的细胞处理条件。
参考文献
产品说明书
预实验

部分蛋白的处理方法,仅供参考

蛋白酶和磷酸酶抑制剂
内源性和外源性的蛋白酶会在细胞或组织裂解后迅速降解蛋白。
在细胞裂解的步骤加入蛋白酶抑制剂混合物可确保蛋白免于降解,磷酸酶抑制剂可以保持蛋白的磷酸化状态。
蛋白酶和磷酸酶的成分组成:

获得高质量的裂解物
根据情况,结合超声、匀浆等物理方法,配合去垢剂裂解。
超声去除裂解样品中DNA干扰,经过超声处理过的样本得到的结果更清晰。
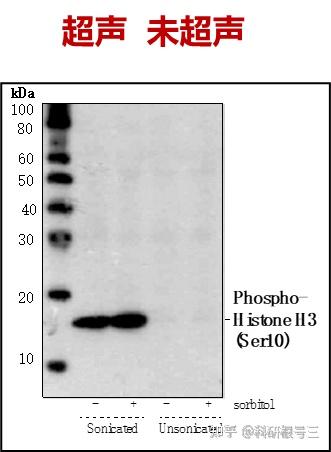
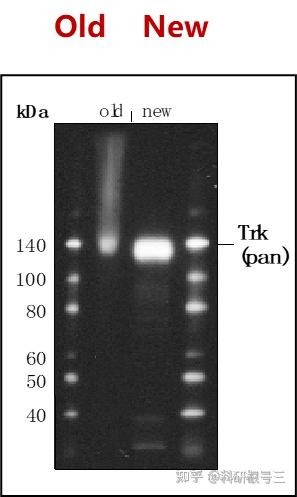
尽量使用新鲜的样品,新鲜的样品制备提取物背景较低。
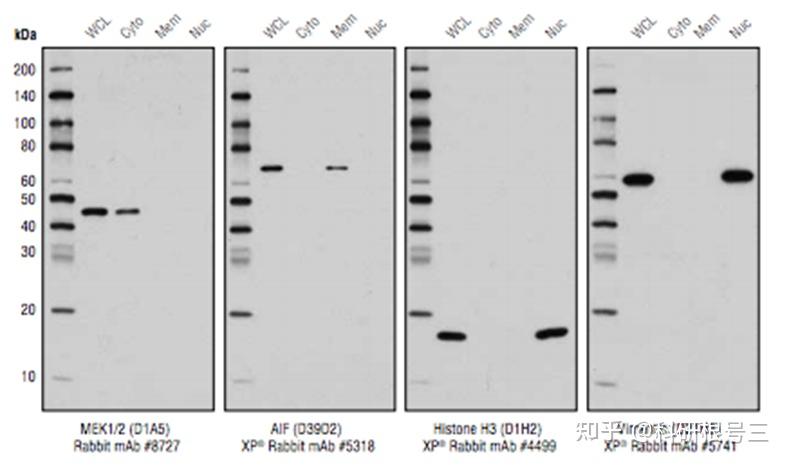
细胞组分分离
细胞组分分离(cell fractionation )是指把细胞的各种细胞器或不同组成成分分离的过程,方便快速的分离细胞组份用于下游分析。
产品推荐:Cell Fractionation Kit #9038

细胞组份分离纯度检测抗体套装,提供一站式解决亚组份分离鉴定方案:
产品推荐:Cell Fractionation Antibody Sampler Kit #11843
不同的亚细胞定位上蛋白的表达不一样,因此在实验前就应该确定目标蛋白的亚细胞定位。
蛋白定量
凝胶电泳时,要求蛋白的上样量尽量一致,后续的结果才更加可信。
为了后续在WB曝光的时候作为内参保持一致的前提。
定量方法

目前主流的蛋白定量方法是BCA检测法,去垢剂兼容性更强。
蛋白浓度测定试剂推荐:
产品推荐:BCA Protein Assay Kit #FMS-W-002
产品推荐:考马斯亮蓝(Bradford)法蛋白定量试剂盒 #FMS-W-001
蛋白变性
普通的SDS-PAGE上样缓冲液组分:SDS、还原剂、甘油、染料以及Tris-HCl(pH6.8)等。
其中最重要的是需要了解溴酚蓝的作用为示踪。

膜蛋白注意事项:
跨膜蛋白有二硫键形成和糖基化等翻译后修饰。请尝试如下条件:
在loading buffer 中加入还原剂,如beta 巯基乙醇,不要煮沸,使用低于70°C的温度,孵育30-60 min。
样品制备的建议

2、凝胶电泳
1)凝胶及电泳液准备
2)上样
3)电泳
主流凝胶:不连续凝胶-浓缩胶/分离胶

在浓缩胶中,其pH环境呈弱酸性,因此甘氨酸解离很少,其在电场的作用下,泳动效率低;而Cl离子却很高,两者之间形成导电性较低的区带,蛋白分子就介于二者之间泳动。由于导电性与电场强度成反比,这一区带便形成了较高的电压梯度,压着蛋白质分子聚集到一起,浓缩为一狭窄的区带。
当样品进入分离胶后,由于胶中pH的增加,呈碱性,甘氨酸大量解离,泳动速率增加,直接紧随氯离子之后,同时由于分离胶孔径的缩小,在电场的作用下,蛋白分子根据其固有的带电性和分子大小进行分离。

如何选择凝胶浓度
凝胶的分辨能力由凝胶的孔径决定,孔径由浓度共同决定。原则有2点:
高比例的凝胶具有较小的孔径,用于分离分子量较低的蛋白。
低比例的凝胶具有较大的孔径,用于分离分子量较大的蛋白。

如何选择凝胶类型?

制备凝胶
pH值正确,妥当的保存,APS需新鲜,现配现用。

保持所有装置清洁。

分离胶勿倒过满,给浓缩胶留一定的距离。

凝胶需缓慢凝固,胶太硬电泳时易烧。

上样
将样本加入到样本孔中


蛋白marker
为了便于观察电泳效果和转膜效果,以及判断蛋白分子量大小,在电泳时通常要使用预染的蛋白分子量标准(蛋白预染Marker)。

产品推荐:彩虹预染marker (5-245 kDa) #FMS-WB003

电泳
蛋白质现在带有负电荷,在电流作用下由负极向正极移动。
产品推荐:Tris-Glycine SDS Running Buffer (10X) #FMS-WB032

电泳仪
电泳仪优势:2膜/4膜、兼容性高、性价比高、配件适配性高。

3、转膜
1)转膜
2)效率验证
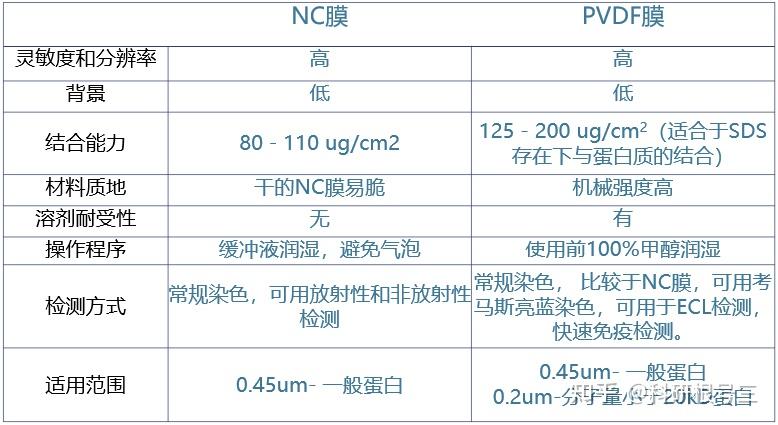
膜的选择

转膜的方法
湿转;半干转;干转


转印仪器

转膜效率验证
带负电荷的丽春红染料能够与膜上带正电的氨基酸发生可逆结合,以判断转膜效率。
CST相关产品推荐:Ponceau S Staining Solution #59803
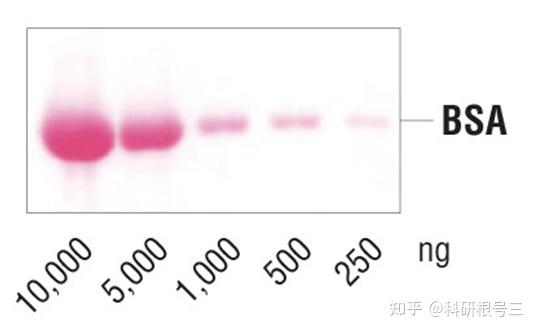
转膜中的要点与建议

4、封闭
转膜完成后要用高蛋白的封闭剂对膜进行封闭,目的是防止抗体和膜发生非特异性结合。
化学发光法:含5% 脱脂奶粉的TBST
荧光法:含5% 脱脂奶粉的TBS
条件:室温封闭1小时
福麦斯相关产品推荐:
脱脂奶粉 #FMS-WB020;
BSA #FMS-WB021;
Animal-Free Blocking Solution (5X) #15019
5、抗体孵育/检测
1)抗体稀释
2)一抗孵育
3)洗涤
4)二抗孵育
5)洗涤
6)检测
稀释缓冲液--依赖于特定的抗体
含5%BSA或5%脱脂奶粉的TBST缓冲液。
说明书确认抗体的最佳稀释液。
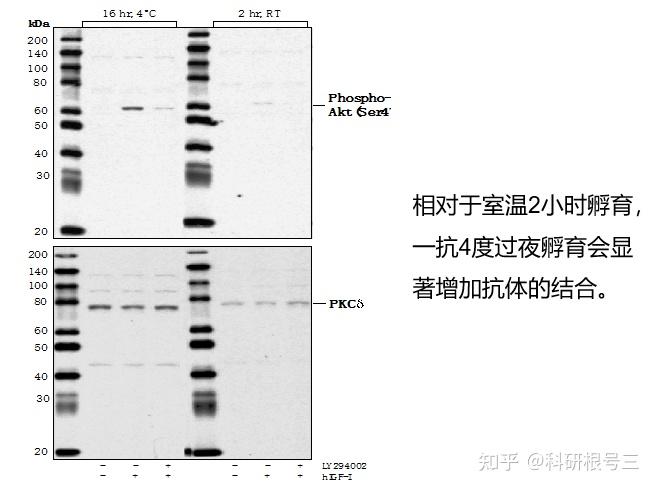
孵育--过夜孵育会改善抗体结合
建议一抗4度孵育过夜。
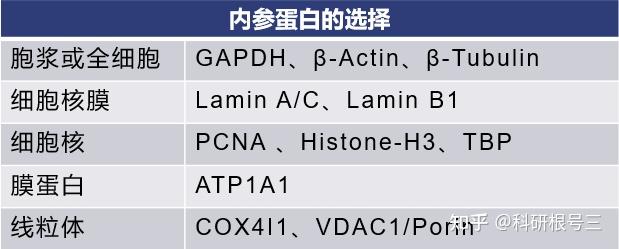
内参蛋白选择原则

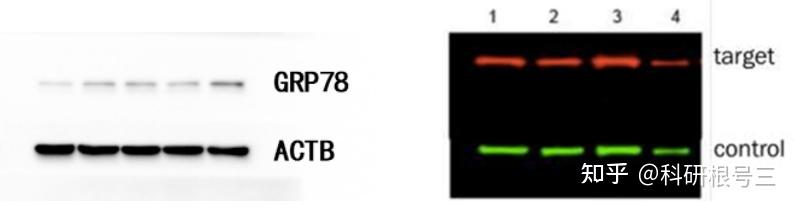
内参蛋白选择指南
注意:没有一种内参蛋白,适用于所有样本的Western Blot,要根据自己的样本选择合适的内参。
洗涤
洗涤缓冲液
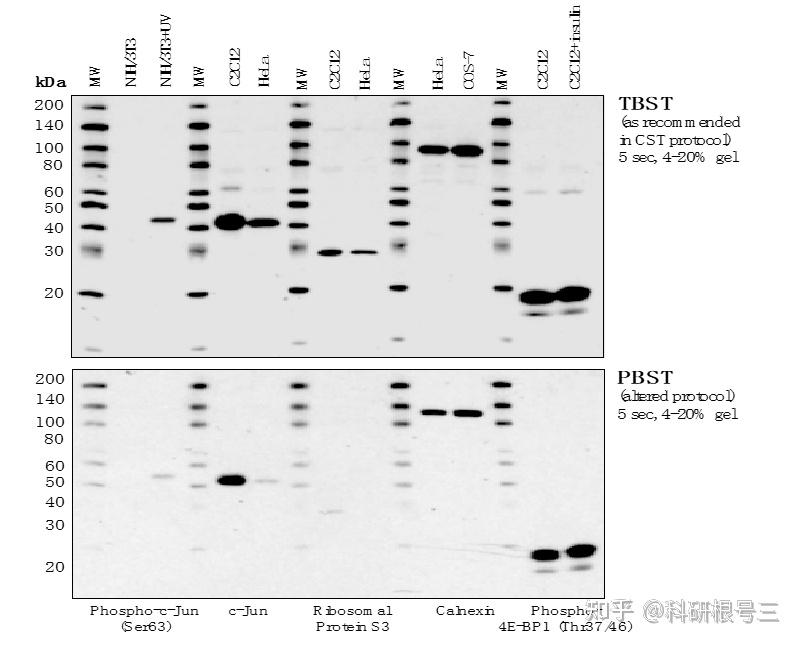
建议使用TBST进行抗体稀释和洗涤。洗涤三次,每次5-10分钟。
对于图中所有抗体,TBST比PBST可以产生更强的信号。
二抗孵育
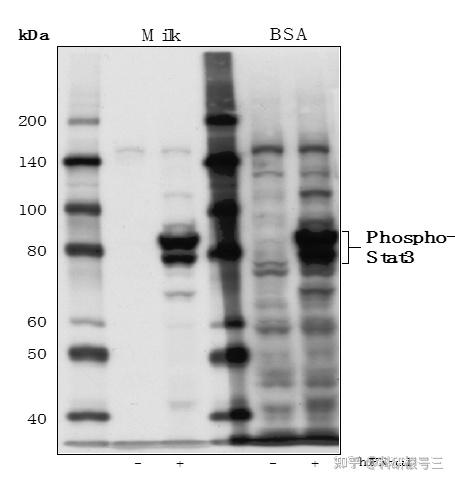
较强的封闭剂稀释二抗可降低背景,建议用含5%脱脂奶粉的TBST稀释二抗。
如下图,用5%脱脂奶粉的TBST稀释二抗比BSA背景更低。
IP后蛋白进行WB实验,可使用构象特异或链型特异二抗,以避免IgG重链或轻链的干扰:
产品推荐:Mouse Anti-rabbit IgG (Light-Chain Specific) (L57A3) mAb #3677;
产品推荐:Mouse Anti-rabbit IgG (Conformation Specific) (L27A9) mAb #3678;
产品推荐:Mouse Anti-rabbit IgG (Conformation Specific) (L27A9) mAb (HRP Conjugate) #5127
检测--ECL发光
化学发光法--市场占用率最广泛的高灵敏度检测方法。
ECL发光液:指一种具有较高的化学发光强度的试剂。
一般使用的发光液A和B,在辣根过氧化物酶(HRP)的催化作用下,A液和B液反应生成一种过氧化物,过氧化物不稳定随即分解,形成一种能发光的电子激发中间体,当后者由激发态返回至基态,就会产生荧光。

WB加速神器--SNAP i.d. 2.0
使用WB加速神器SNAP i.d. 2.0,30分钟可完成实验。

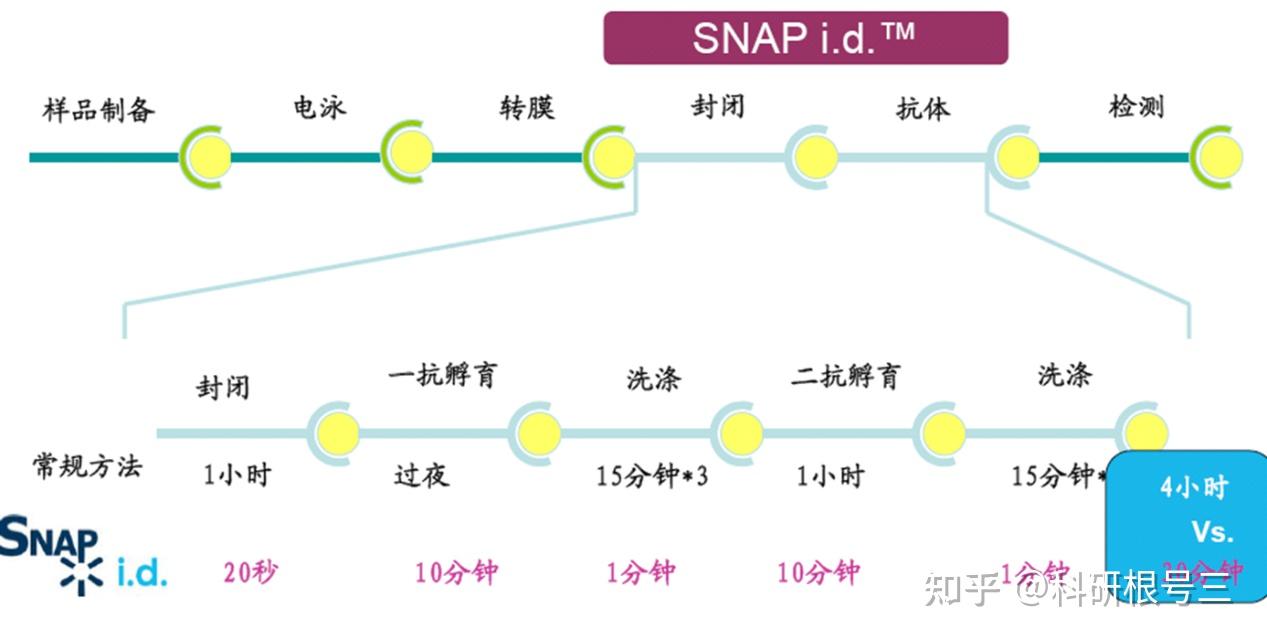
Snap i.d. 原理
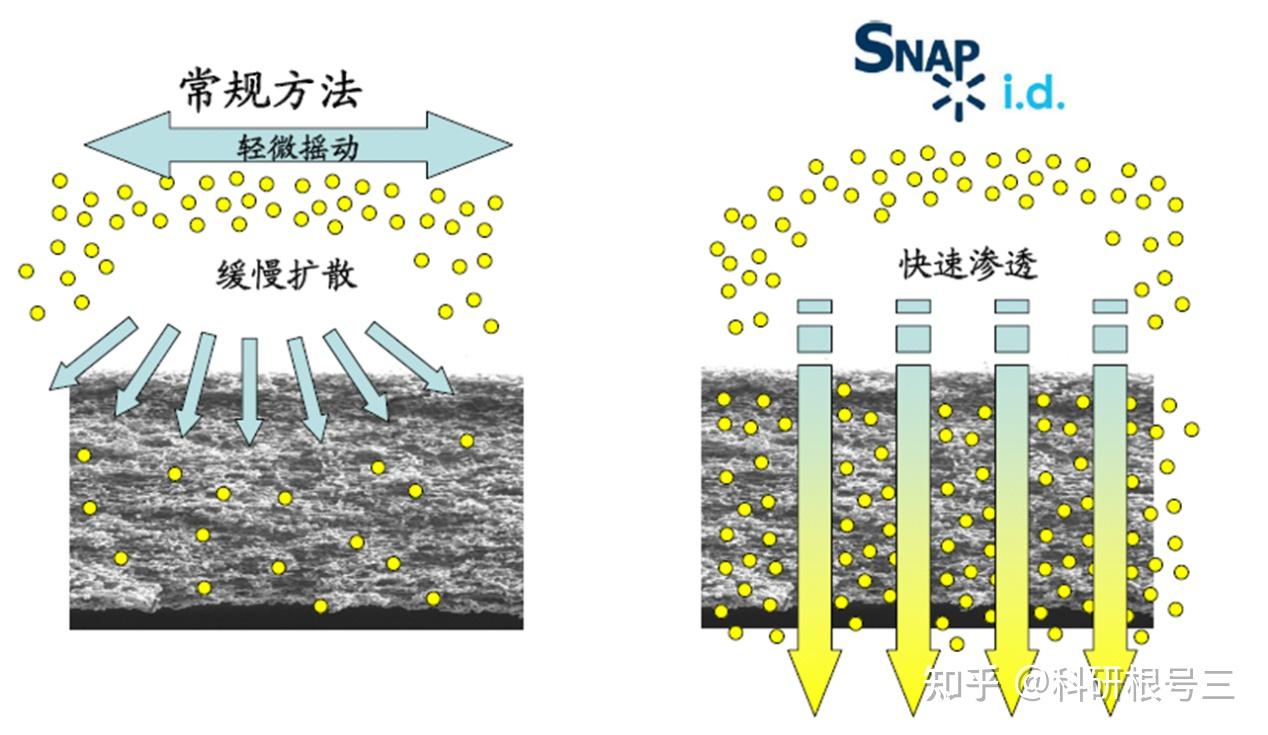
Snap i.d. 与传统方法的实验结果对比

二、Western Blotting检测的操作方法和注意事项






三、Western Blotting 实验问题及解决方案
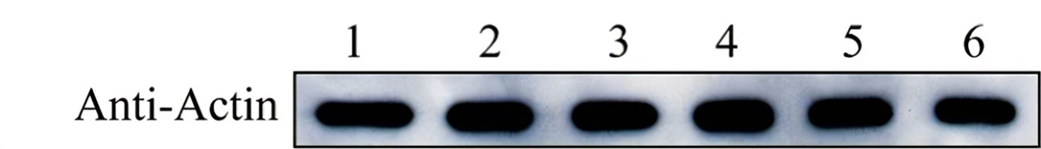
正常条带示例
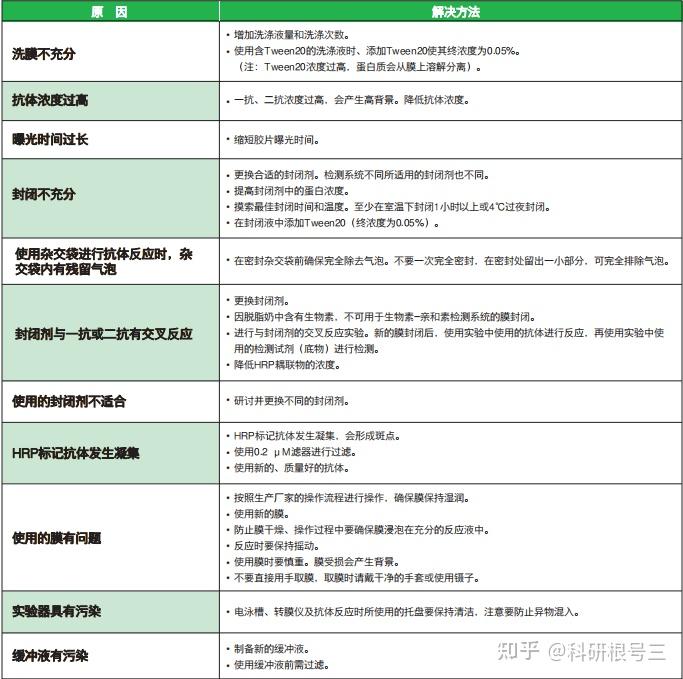
问题1:有污渍或斑点,整体背景普遍偏高
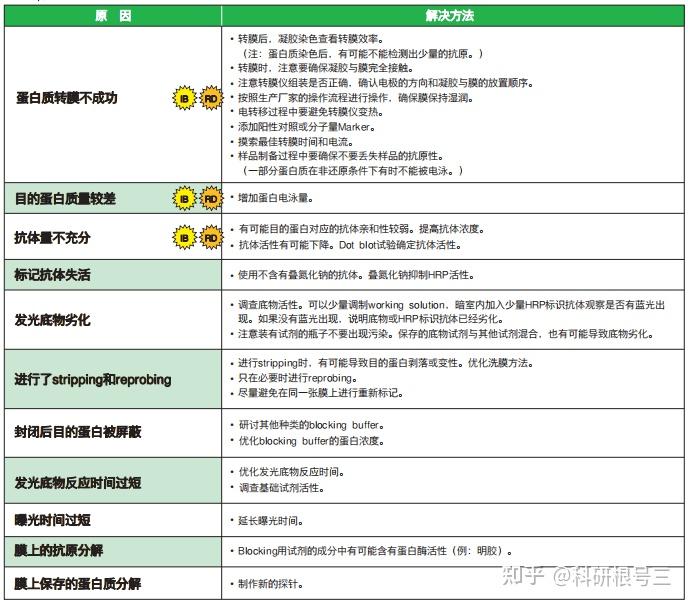
问题2:出现白色条带

问题3:出现非特异性条带

问题4:信号弱或无信号
Western BLoT Immuno Booster(制品code T7111A)可有效改善结果。
Western BLoT Rapid Detect v2.0(制品code T7122A) 可有效改善结果。
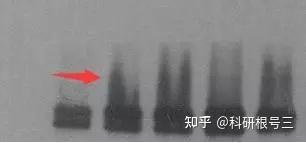
问题5:膜上部分无信号

问题6:条带扩散

四、Western Blot中结果条带的各种稀奇古怪结果图解析
1. 啥也没有

原因:比较多,如果单纯一张没有任何显色的X光片,最可能是一抗加成其他抗体,或者二抗种属加错了,比如兔的加成鼠的。
解决办法:仔细检查抗体是否加错,确认转膜没有问题。
经验:上面的图片展示的是一点信号都没有,如果是这样大部分情况是抗体加错了。如果中间出现了细微的条带,可能原因是蛋白上样量太少,一抗浓度过低,ECL发光液失效。另外如果转膜出现了问题,比如膜放反了,自然是一个白片。
2. 高背景

原因:封闭不够好,一抗浓度高,洗膜时间和次数不够
解决办法:降低一抗浓度,增加洗膜时间和次数。
经验:高背景可能是WB中最常见出现的问题,目的条带单一清晰,但是其他地方又弥漫性较为均一的背景(比较连续的)。其实只要我们注意操作规范,不偷工减料就很容易避免,洗膜按照规定来5min*5次或者10min*3次,不要改成5min*3次,或者10min*2次。
3. 非特异性条带

原因:一抗非特异性与蛋白结合
解决办法:更换一抗
经验:此种情况绝大多数是因为一抗不好,你无法判断那一条是目的条带。如果实在没有更好的抗体,建议采用阴性对照和阳性对照来确定上述哪个条带是目的条带。当然这种情况下有一种很小几率的可能是一抗浓度太高引起的非特异性结合。
4. 条带中出现边缘规则的白圈

原因:电转中膜和胶之间存在气泡。
解决办法:转膜前去掉膜和胶之间的气泡
经验:我们常常将电转液倒入一个盘子里,倒入的液体不能太多也不能太少,最好的高度是与放上第一层滤纸齐平,然后往滤纸上浇点转膜液,把电泳胶用清水清洗下,将电泳胶平铺到滤纸上,仔细检查滤纸与胶之间是否有气泡,可以左右前后观察,不同方向观察之后确认无气泡,然后再往胶上面浇点电转液,用两只手的拇指和食指轻轻夹住PVDF膜的两侧中间,使膜成U型,然后将U型的底部接触胶的中间,慢慢往两边放下膜,这样一般气泡很少。然后上层滤纸同样用U型的放置方法,用玻璃棒稍微贴实下,然后盖上海绵。注意不要来回赶气泡,这样反而会带入气泡。
5. 条带中间出现白色(反白)

原因:中心部位高浓度HRP把底物消耗过快,中间部位底物消耗结束之后就不发光了
解决办法:降低蛋白量,降低一抗和二抗的浓度。
经验:如果你足够迅速,可以在中间部位底物消耗之前就把X光片给定影出来,但是时间很难把握,建议还是从降低蛋白量,降低一抗二抗浓度入手。
6. 出现黑点和黑斑

原因:膜上其他部位与一抗或者二抗非特异性结合
解决办法:封闭牛奶一定要纯,封闭结束之后要洗
经验:我们常常是等到要封闭的时候发现没有牛奶,然后匆匆忙忙用PBST/TBST配置,配的匆忙的时候往往不管是否已经完全溶解,如果没有完全溶解的情况下加牛奶倒膜上,会导致很多不溶性颗粒附着在膜上,这就会导致发光时候膜上的黑点。所以牛奶溶解之后,最好静止一下,然后轻轻地吸取上层牛奶进行封闭,封闭结束之后一定要洗三遍之后再加一抗。
7. 条带拖尾
原因:蛋白量太大,一抗浓度和时间太长
解决办法:根据情况调整蛋白量,同时一抗浓度和时间也可以缩短。
经验:这种情况很容易出现,因为很多原因都可能导致这一个结果。一般来说,蛋白量都是我们经过很多次摸索得出的最适蛋白量,因此不太可能是蛋白量过多引起的。最有可能的是因为一抗浓度太高,作用时间太长引起的。另外洗一抗和洗二抗千万不要偷工减漏,建议5min*5次,不要但是洗这么多次就把抗体和蛋白洗掉了,你又不是拿刀在上面刮,真正的抗原抗体的结合是通过这种方式洗不掉的。
8. 出现重影

原因:荧光强度比较高,在压片时,放好之后不小心又轻微移动了一段距离
解决办法:X光片放上去之后,就不要动了,即使放歪了也没关系。
经验:有的时候出现重新刚好在上下位置,并且重影会相对弱一些,不要误以为抗体识别了该蛋白的另外一种异构形式。
9. 出现非均一性背景

原因:膜可能曾经干过
解决办法:在每一步的操作过程中,都需要注意不要让膜干
经验:在封闭的时候,洗一抗,洗二抗,以及发光的时候都时刻需要注意蛋白面不要风干,风干之后结果很可能就是这个样子。注意与高背景区别。
10. 某个条带变形

原因:SDSPAGE胶中存在气泡或者某不溶性颗粒
解决办法:配胶过程中要小心,使用无杂质的液体。
经验:很多实验室中使用的不是最新的设备,比如配胶用的海绵垫,如果用了很多年之后,会从下面往上面漏小气泡,当气泡足够小并且胶快凝固的时候,走到中间的小气泡就停留在胶内,并会影响到后面的跑胶。另外配胶用的水,SDS,Tris缓冲液要注意不要有杂质。
11. 条带呈哑铃状

原因:配置胶有问题
解决办法:把胶配好,不合格的胶坚决不用
经验:出现哑铃最大的可能是胶没有配置好,胶凝固后不均一,不知道大家有没有出现如下的配胶情况,下图中示意拔完梳子之后的结果,如果你拔完梳子之后出现图中下面部分的样子,多半会出现哑铃状。另外还有一种可能是样品中含有太多杂质,没有离心下来,然后杂质沉积在孔的中间,蛋白自然被推挤到两边。
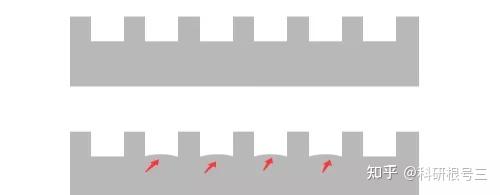
12. 最边缘条带弯曲

原因:电泳电流不均一
解决办法:换用新的电泳槽; 不使用两边的两孔
经验:一般我们使用的是10孔的的胶,如果你上样刚好10个孔,那么最两头的两个孔肯定会歪曲。另外上样最好在胶的中间,这样电场均一。
13. 其他问题
(1)蛋白分子量偏高或者偏低。可能是胶的浓度与目的蛋白的浓度不对应,比如说100KD的蛋白你用12%的胶跑,或者说20KD的蛋白你用6%的胶跑。
(2)蛋白质降解。蛋白质降解后很可能会在比原来位置低的地方出现主带,然后会出现一些其他带,最主要特点是所有的条带比正常的都低,并且条带模糊不清晰。
(3)所有条带连成一片没有间隔。原因最可能是上样量过多,其次是样品弥散(比如电泳长时间停止样品弥散)。
14. 电泳过程中出现现象及问题
(1)整个条带呈 “ ︶ ” 状:凝胶冷却不均一,电泳槽老化。
(2)整个条带呈“ ︵ ”: 凝胶左右两头没有凝固好
(3)溴酚蓝拖尾:样品溶解不好。
(4)纵向的纹理:上样样品中存在不溶性颗粒
(5)溴酚蓝很粗:浓缩胶浓缩效果不好,可能是浓缩胶太短,或者是浓缩胶配错。
(6)在分离胶中跑不动:Tris-Cl PH值不对,或者忘记加SDS。
有时间再把一些经验分享给大家,今天就先到这里~
点个赞同,祝你实验顺风顺水~

推荐内容
绝密!2023年国自然立项清单表可下载(生命科学部+医学科学部),附往年1000+份中标标书!
如何查看国家自然科学基金的的摘要和下载结题报告?
有哪些好用的zotero插件?
有什么适合药学/生物方向科研小白的简单易学的科研绘图工具?
有没有什么很好的科研作图软件?
顶级图像分析软件,Image J、Fiji、Image pro plus,一次帮你搞定!
NoteExpress和EndNote相比哪一个更好用?(附安装包)
2023年更新!EndNote X9永久激活版本(序列号激活),最稳定,可汉化!
写的太全了,Endnote 插入文献以及调整参考文献格式,这是知乎上最好的教程!
太牛了!Zlibrary回归,这里有最稳定的访问指南!
western blot是为了什么?
如何学习和记忆细胞信号通路?
最值得推荐装!GraphPad Prism 9.3 ,功能更多!
SPSS 27 安装激活:授权到期时间2037年
流式数据分析标杆神器,FlowJo 10 分享!
 新公网安备 65010402001845号
新公网安备 65010402001845号